
SITE MAP
-
Provider
-
Patient
-
How to order
-
Other Test
-
Contact us

Test options
Provider
- Home
- Provider
- Test options
- Oncocatchㆍs
- Oncocatch ㆍe
- Oncocatch ㆍcdxs
01
Methylation-based Liquid Biopsy Screening:
Highly sensitive, Highly scalable, Clinically validated, Localization of Tumor
OncoCatch-E uses methylation-sensitive restriction enzyme digestion followed by sequencing (MRE-seq) using cell-free DNA (cfDNA) for the diagnosis of cancer and tissue-of-origin (TOO) using a deep neural network (DNN). Among the genomic features in blood, methylation patterns could be useful not only for determining whether cancer is present but also for revealing its origin. The overall accuracy of OncoCatch-E is high because MRE-seq measures global hypomethylation in the cancer genome. OncoCatch-E shows 78.1% and 66.3% sensitivity at 99.2% specificity for colorectal cancer and lung cancer respectively. The receiver operating characteristic curve (AUC) is 0.978 for colorectal cancer and 0.956 for lung cancer. The true-positive rates of the TOO model is 94.4% and 89.9% for colorectal and lung cancers, respectively.

02
Background
Liquid biopsy tests for the presence of circulating tumor cells or cancer-cell-derived nucleic acids, e.g., cell- free DNA (cfDNA) or regulatory RNAs (e.g., miRNA) in blood or urine. Tissue biopsy is performed only on the relevant tissue after signs of cancer are detected, whereas liquid biopsy has the advantages of enabling rapid diagnosis through minimal invasiveness and obtaining representative information of the whole tumor cell population.12 cfDNA is a crucial biomarker in liquid biopsy and has been studied extensively for the early detection and prediction of cancer prognosis. 3-5 Recently, research for detecting cancer-specific methylation signals has been conducted.6- Cancer-specific DNA methylation patterns occur when normal cells are transformed into cancer cells, with each cancer type having unique methylation characteristics 9-10 Therefore, detecting methylation could confirm the existence of cancer or distinguish cancer types by detecting the cancer-specific DNA methylation pattern from cfDNA in blood. 12 MRE-seq, which cuts and captures specific unmethylated sequences using a methylation-sensitive restriction enzyme, is a high-potential alternative approach, but it has not been studied for liquid biopsy.34 MRE-seq has two major advantages as a liquid biopsy method. First, it can analyze global hypomethylation, a characteristic of cancer genomes.15.16 It selectively cuts and sequences unmethylated restriction sites of DNA, which enriches ctDNA, increasing sensitivity. Second, MRE-seq degrades DNA less during library preparation compared to bisulfite conversion; thus, accurate analysis is possible using a relatively small amount of cfDNA.1738
03
Methods
Data are presented as numbers (36) or the median (interquartile range).
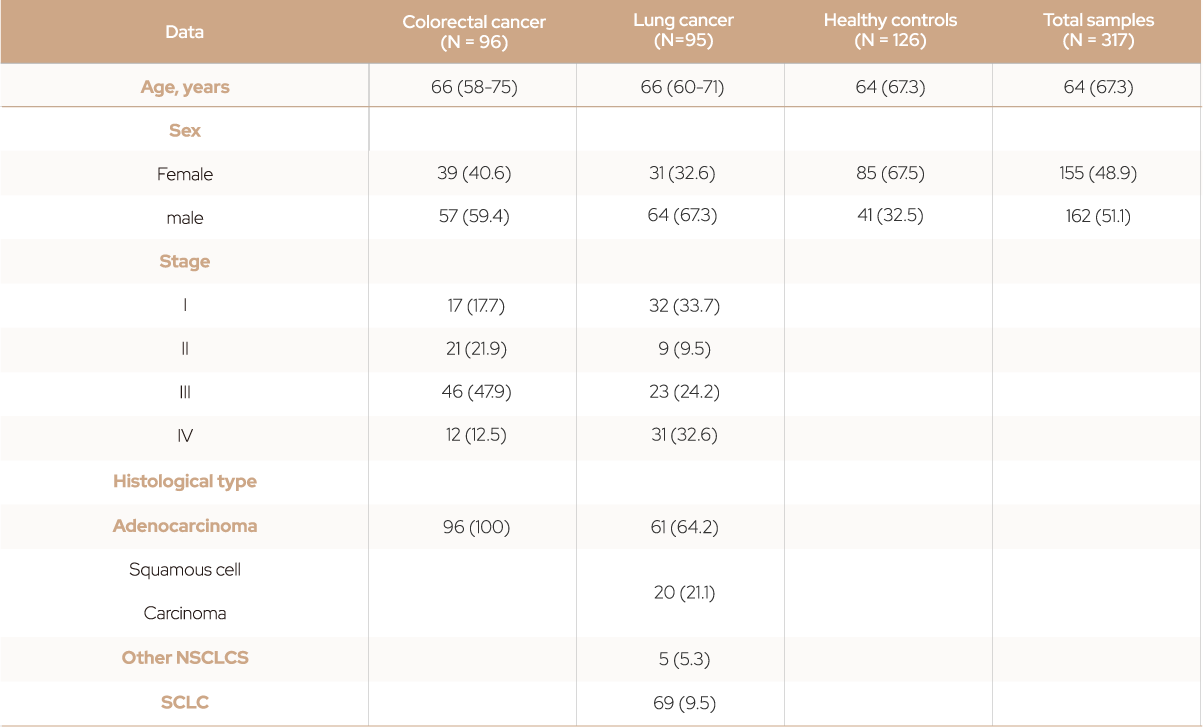
Table 1. Demographics of patients with cancer and healthy controls.
03-1
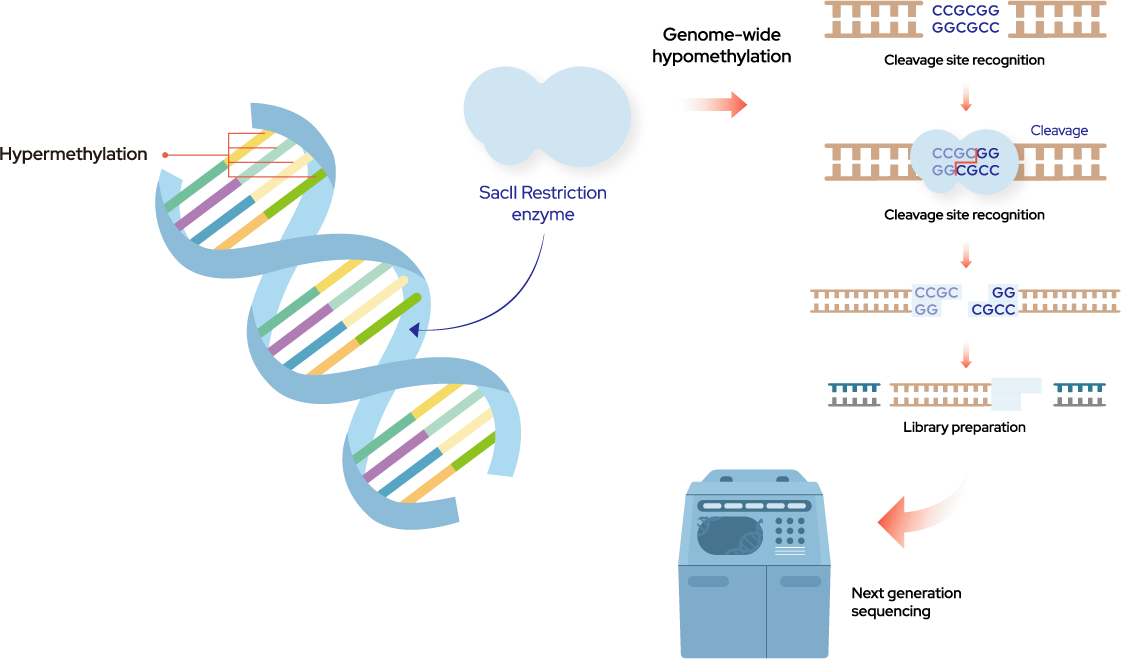
Figure 1. Next-generation sequencing (NGS) of MRE-seq and Downstream analysis
10 mL of blood was collected from the cancer patients and healthy donor into cfDNA Vangenes Cell Free DNA Tubes (Vangenes, US) and centrifuged at 1900 g for 15 min at RT. The plasma was transferred into 1.5 mL tubes and then centrifuged at 16,000 g for 15 min at RT. cfDNA was extracted from 4ml plasma with chemagen 360 which was prepped to library with OncoCatch E kit. Respective libraries were pooled and sequenced on Illumina NovaSeq 6000 (Illumina) with 100 cycles kit with paired end reads. Fastq data were aligned to the hg19 human genome reference after quality trimming. Polymerase chain reaction (PCR) duplicated reads were removed using unique molecular identifier (UMI), and the on-target reads in Sacll sites were normalized to proceed with the downstream analysis and deep neural network (DNN) training.
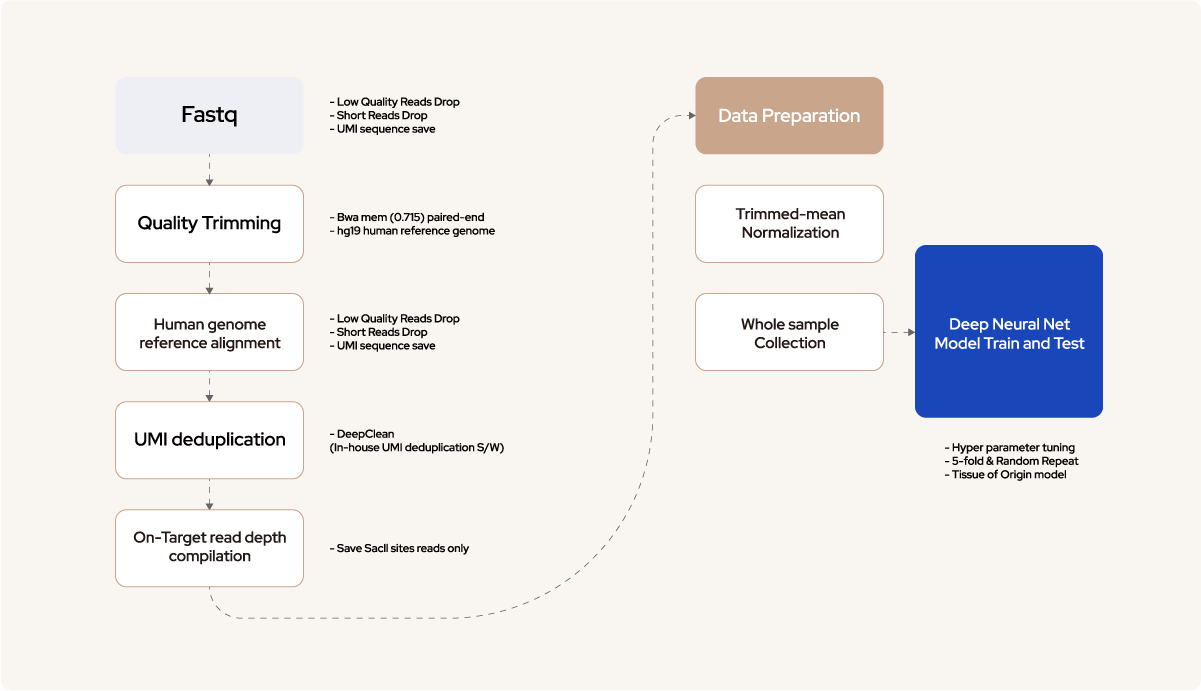
04
Deep learning modeling
and Performance assessment
04-1
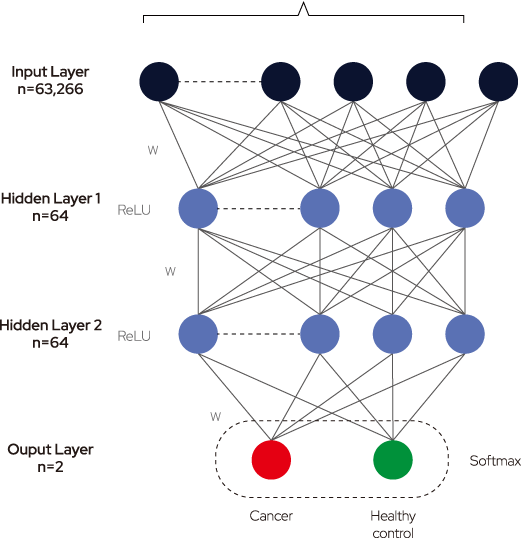
Figure 2. Deep neural networks (DNN) architecture and Performance evaluation
DNN consists of an input layer, an output layer, and many hidden layers in the middle. Each layer consists of neurons that have a weight value and the activation function. During the training process, each weight value is updated using a gradient descent algorithm andbackpropagation. The normalized depth values corresponding to the 63,266 Sacll sites entered the input layer and went through two hidden layers. Training was accomplished by calculating the loss of the cost function in the final output layer. The weight value of each node was updated by calculating the cross-entropy loss through the SoftMax activation function of the output layer and by performing backpropagation by 120 epochs toward a decreased value. To build an accurate and robust prediction model, the dataset was split into training, testing, and validation sets. The training set encompassed the data sample used to fit the model, whereas the validation set was used to fine-tune the hyperparameters, ie, the number of layers and nodes and batch and epoch sizes. The model was trained with the best parameters, and the test dataset was evaluated.
04-2
Sacil Digestion Sites (63,266)
04-3
Performance evaluation strategy
05
Results
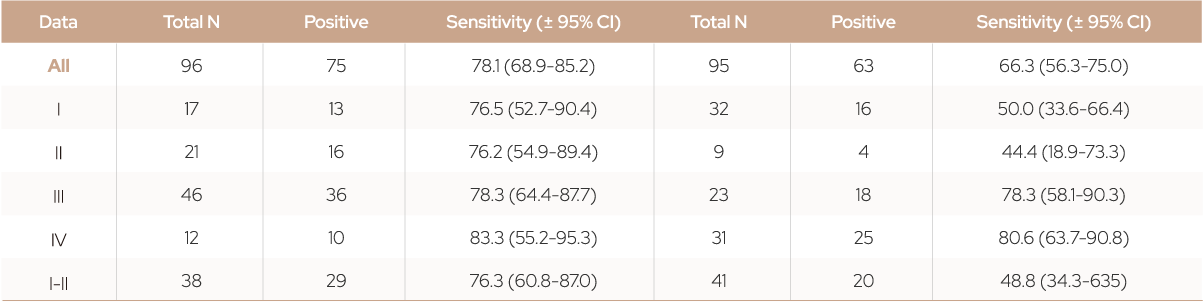
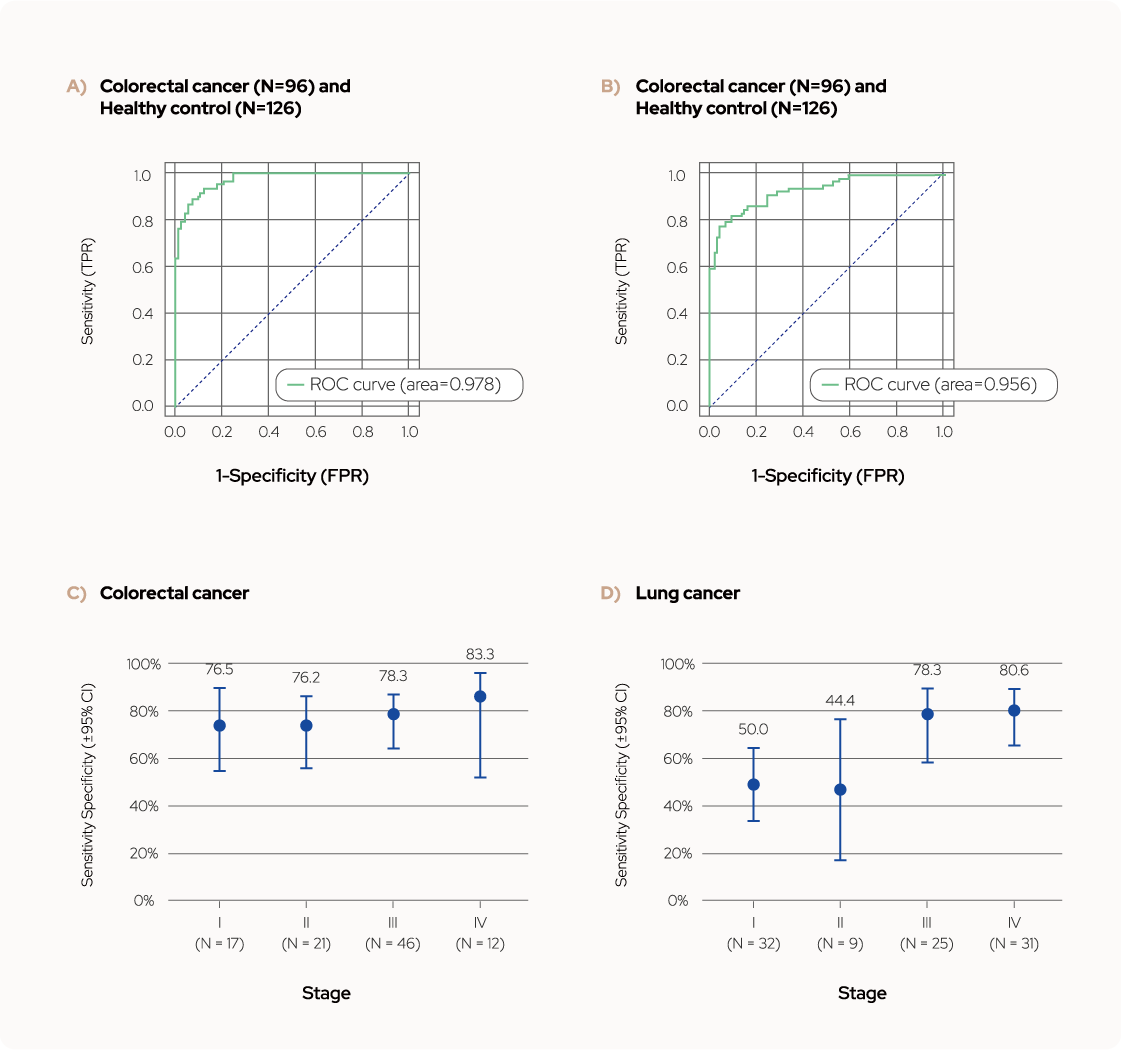
Figure 3. Test performance of Colorectal cancer and Lung cancer model.
(a, b) The overall AUC values were 0.978 for colorectal cancer and 0.956 for lung cancer. (c, d) Sensitivity at
99.2% specificity with 95% confidence interval (CI) according to cancer stage.
06
TOO (Tissue-of-Origin)
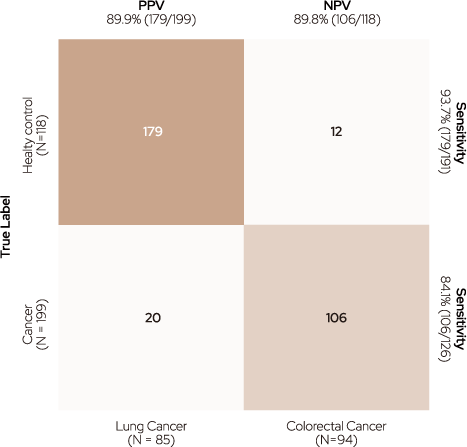
TOO was implemented with two models: the Cancer Classifier, which determines whether cancer is present, and the Cancer Type Classifier, which classifies the type of cancer. According to the Cancer Type Classifier, cancer samples were positively predicted with a sensitivity of 93.7% based on a threshold of 0.5. Moreover, 179 samples with true-positive cancers were accurately classified into the two cancer types using the Cancer Type Classifier (94.4% in colorectal and 89.9% in lung cancer).
Colorectal Cancer
Cancer type classifier
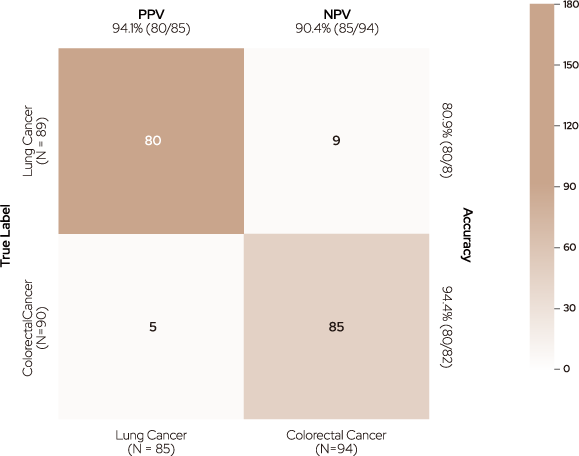
Predicted Label
Figure 3. Confusion matrix of tissue-of-origin (TOO).
TOO accuracy was measured using two different classifiers : the Cancer Classifier for determining cancer existence and the Cancer Type Classifier
for identifying the type of cancer.
References
1. Cheung AH-K, Chow C, To K-F. Latest development of liquid biopsy. J Thorac Dis. 2018 10:51645-51
2. Wan JCM, Massie C, Garcia-Corbacho J et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 201717-223-38.
3. Hu Z, Chen H, Long Y et al. The main sources of circulating cell-free DNA: apoptosis, necrosis and active secretion. Crit Rev Oncol Hematol. 2021,157:103166.
4. Gale D, Lawson ARJ, Howarth K et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS One. 2018,13:0194630
5. Huang C-C, Du M, Wang L. Bioinformatics analysis for circulating cell-free DNA in cancer. Cancer. 2012;11:805.
6. Li W, Zhou XJ. Methylation extends the reach of liquid biopsy in cancer detection. Nat Rev Clin Oncol. 2020:17:655-6
7. Constancio V, Nunes SP, Henrique Retal DNA methylation-based testing in liquid biopsies as detection and prognostic biomarkers for the four major cancer types. Cell. 20209:624.
8. Liu MC, Oxnard GR, Klein EA et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020,31:745-59
9. Hao X, Luo H, Krawczyk M et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci USA. 2017;114:3414-9.
10. Yang X, Gao L, Zhang S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief Bioinform 20178:761-73.
11. Barefoot ME, Loyfer N, Kilit AJ et al. Detection of cell types contributing to cancer from circulating, cell-free methylated DNA. Front Genet. 2021:12:671057.
12. Zhang C, Xu R. Predicting cancer origins with a DNA methylation-based deep neural network model. PLoS One. 2000;15:0226-461.
13. Xing X, Zhang B, Li D et al. Comprehensive whole DNA methylome analysis by integrating MeDIP-seq and MRE-seq. In Tost J (ed): DNA Methylation Protocols. New York, NY: Springer 2018-209-246. (Methods in Molecular Biology:vol 1708).
14. Galardi F, De Luca F, Romagnoli D et al. Cell-free DNA-methylation-based methods and applications in oncology. Biomolecules. 2020:10:1677.
15. Ansar M, Wang C-J, Wang Y-H et al. SMADS hypomethylation as a biomarker for early prediction of colorectal cancer. Int J Mol Sci. 2020:21:7395.
16. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1239-59.
17. Huang J, Wang L. Cell-free DNA methylation profiling analysis-technologies and bioinformatics. Cancers. 2019,1341.
18. Grunau C. Clark SJ, Rosenthal A. Bisulfite genomic sequencing systematic investigation of critical experimental parameters.Nucleic Acids Res. 200129:£65-65


#291 Harmony-ro, Yeonsu-gu, Incheon, 22014, South Korea
EDGE | Name of Representative : Min-Seob Lee
Tel : +82-32-713-2100 | Email : info@edgc.com | Business Registration Number : 131-86-48582
EDGC, Inc. | #143, Gaetbeol-ro, Yeonsu-gu, Incheon, 21999, South Korea | Tel : +1-800-605-8422
ⓒ[2023]. Eone Diagnomics Genome Center Co., Ltd. All Rights Reserved.